
How Technology Has Changed Computer System Validation
By Richie Siconolfi, Richard M. Siconolfi, LLC

AI, blockchain, the list goes on. For those of us who research, develop, test, submit, and release regulated products to the public, these continuous technological advances are a double-edged sword. Yes, the computerized systems, equipment, and instrumentation have allowed us to increase the speed of research and development; however, we also have to prove that all of those computerized systems, equipment, and instrumentation meet regulatory requirements. This is a daunting, but not impossible, task.
This three-part series will explore the use of computerized system compliance in clinical research beginning with a primer on two things: the predicate rules (regulations), guidance documents, directives, and guidelines; and the scientific method. Both impact the way we protect public health and safety — so much so that the major regulatory agencies, such as the FDA, EMA, International Conference on Harmonization (ICH), and U.K. Medicines and Healthcare Products Regulatory Agency (MHRA), have provided us with enough regulations and guidance documents to make us dizzy. Thankfully, they are also the first to offer help in reaching compliance. Herein, I’ll introduce the concepts of a computerized system and the scientific method, discussing how the two intersect in the pursuit of computer system validation.
What Is A Computerized System?
To start, we must first define what a computerized system is. The FDA’s Glossary of Computerized System and Software Development Terminology defines a computerized system as one that “[i]ncludes hardware, software, peripheral devices, personnel, and documentation; e.g., manuals and Standard Operating Procedures1”. This definition also references computer systems, which are defined by the American National Standards Institute (ANSI) as “a functional unit, consisting of one or more computers and associated peripheral input and output devices, and associated software, that uses common storage for all or part of a program and also for all or part of the data necessary for the execution of the program; executes user-written or user-designated programs; performs user-designated data manipulation, including arithmetic operations and logic operations; and that can execute programs that modify themselves during their execution. A computer system may be a stand-alone unit or may consist of several interconnected units.2”
Part 11, The Electronic Records & Signature Rule
There have been many regulations that have changed how we approach computer system validation, and some of us have even launched careers in regulatory compliance. The one regulation that has had the biggest impact on computer system validation in my opinion is the rule on electronic records and electronic signatures promulgated by the FDA in 19972 (affectionately called Part 11). Why? It was the first FDA regulation to state that if you decide to electronically create, modify, maintain, archive, retrieve, or transmit records under any requirements outlined in agency regulations, i.e., predicate rules, you must comply with Part 11. This regulation put an entirely new perspective on computerized systems, electronic records, electronic signatures, and computer system validation.
The Scientific Method
The scientific method, with roots in the Ancient Greeks and other early civilizations, requires the scientific investigator to follow these six steps:4
- Observation
- Question
- Hypothesis
- Experiment
- Results
- Conclusion
The importance of the method is the structure itself, i.e., how science is documented. There were many early documents on the scientific method, many describing how to document scientific experimentation per discipline. Instead, let’s discuss documenting science as we know it today. Early scientific investigations were recorded on paper in bound laboratory notebooks and later preprinted paper templates, which may have been taped into bound laboratory notebooks or placed in three-ring loose sheet notebooks. As with all types of notebook entries, some initial training was required to accurately record observations, pose questions, and develop a hypothesis. These manual operations were the mainstay of scientific documentation for many years. Then came computing devices that allowed us to record experimental results directly into a database where we would analyze the data to produce results based to prove or disprove our hypothesis faster. The results of our experimentation may have produced another observation and the cycle would have started again.
The structure of the scientific method is similar to the software development life cycle (SDLC) that many software vendors and regulated companies use today. The Society of Quality Assurance presented this traditional model of an SDLC3:
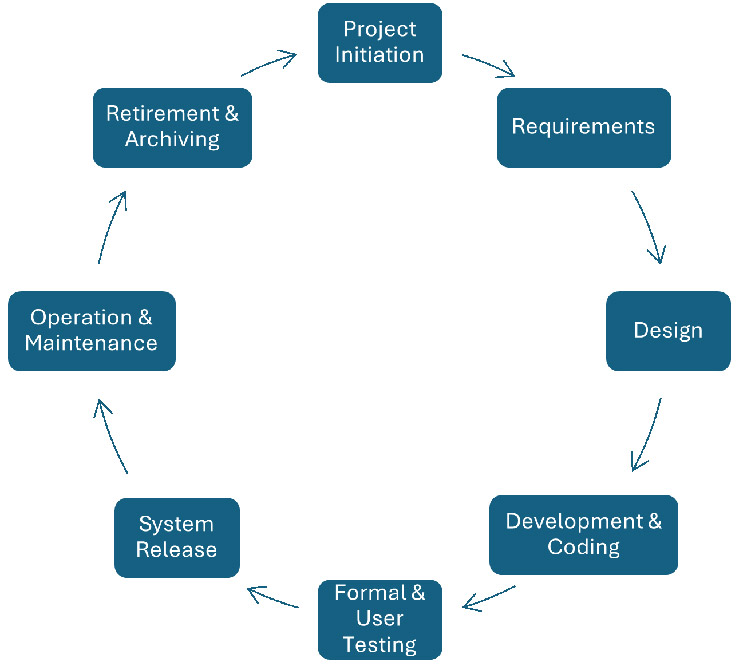
Figure 1: Software development life cycle
While the number of phases between these two methodologies may differ, the processes are similar. For example:
- The Scientific Method starts with Observations. Observations can equate to requirements, and Requirements are what the users of a computerized system need to automate or streamline a function using a computerized system.
- Question and design align because the question is the first step in forming a hypothesis. Design and Coding are the activities of building a computerized system to meet the needs of the user’s specific functions.
- Experiment is linked to Formal & User Testing. An experiment is designed to prove or disprove a hypothesis. Testing provides evidence that the computerized system is functioning as designed, i.e., proving the design and coding were meeting the users' requirements.
- Results and System Release: Results of the experiment either prove or disprove the hypothesis. A system is not released unless the testing meets its acceptance criteria. If the testing fails, it is the same as disproving the hypothesis and then coding may have to be revised, followed by additional testing.
- Conclusion and Maintaining the Validated State: Both attributes have the same goal. The conclusion is the scientist’s interpretation of the results vs. the hypothesis. Did the data support the hypothesis? If yes, does the conclusion generate another question? And the process continues. If not, then the scientist revises the hypothesis again and conducts another experiment, etc. Maintaining the Validated State is a bit different. Maintaining the validated state of a computerized system is to ensure the system functions as intended. However, when new requirements are demanded by the user, or there is a bug, the process starts again.
Evolution Of Technology
Some of the early tools that assisted us in analyzing our results were slide rules, simple handheld calculators, programmable calculators, and personal computers. Early IBM mainframe computers required punch cards or paper tapes, followed by magnetic tapes and floppy discs. Then came the era of web-based computer programming, followed by software as a service (SaaS) and cloud computing. Each step in this technological transformation required tighter controls to guarantee security, data quality, and data integrity. Both paper documentation and electronic records have their advantages and disadvantages and are still used today. The point is that technology continues to move forward at its own pace. This pace will always challenge us to rethink how we need to validate our computerized systems.
How To Approach Computer System Validation
The FDA has stated that validation is “[e]stablishing documented evidence which provides a high degree of assurance that a specific process will consistently produce a product meeting its predetermined specifications and quality attributes.”2
The FDA definition focused on these three attributes:
- Documented evidence
- High degree of assurance
- Quality attributes
Computer system validation has been around for a long time, and as stated above the technology and the interpretation of government agency regulations and guidance documents may have changed the way we look at validation because of the speed at which technological advancements occur. There are two other FDA government guidance documents we should consider. The first is the FDA Part 11 guidance document on scope and application5. It was published in 2003 after the regulated industry stated that some Part 11 guidance documents were impeding or confusing compliance. The FDA decided to withdraw almost all of those guidance documents. The FDA’s scope and application guidance document did three things: it narrowed the scope of the regulation; it allowed enforcement discretion for audit trails, validation, and record retention; and it recommended that the industry adopt a risk-based approach to computerized system validation.
This allowed the industry to develop specific risk models using critical thinking and enforcement discretion to research, develop, and test their new risk-based approach to computer system validation. It should be noted that the FDA did not rescind Part 11; it only stated that it would apply enforcement discretion to audit trials, validation, and record retention. All of the remaining Part 11 requirements must still be followed.
Each regulated company, through critical thinking, refined their risk-based approach and developed a Part 11 assessment document (i.e., their risk-based approach to computer system validation). The regulated industry revised its standard operating procedures on validation and updated its validation deliverables. This was not an easy or quick process. From my point of view, interpreting the scope and application guidance document required appointing team members from those who are experienced with the Good Laboratory Practice, Good Clinical Practice, and Good Manufacturing Regulations by regulated authorities around the world and experts in data management, IT, and regulatory affairs to develop and test risk-based approaches. The results allowed the various departments to better understand validation by understanding the risk levels they assigned to their computerized systems using an agreed-upon risk-based approach. Simply put, the high-risk computerized system followed the traditional path of validation. Medium- and low-risk computerized systems required less documentation and testing. This led to the publication of our methodology by DIA in 20076.
It has taken 18 years, but the FDA has issued another draft guidance document on computer system validation, “Computer Software Assurance (CSA) for Production and Quality System Software”7. This draft guidance document picks up where the scope and application guidance document left off. This draft guidance recommends reviewing your current risk-based approach, and
- adjusting risk levels,
- allowing for scenario and unscripted user acceptance testing, and
- revising validation documentation to reflect the opportunities listed in the draft guidance appendices.
While we are still waiting for the publication of the final CSA guidance document, many in the regulated industry have already implemented CSA programs within their CSV methodology.
Summary
Technology will continue to challenge how we interpret and apply regulations and guidance documents we rely on for regulatory compliance. Improvements in computerized systems, equipment, instrumentation, and how we store data will continue to evolve. Computer system validation models and the addition of CSA into these models will reinforce the idea that a documented risk-based approach to validation will continue to streamline the process of computer system validation. The ultimate goal is to ensure a computerized system has been designed and tested for its intended purpose.
Up Next
The next article in this series will focus on the interface between regulated product lifecycle and technology, starting with the dawn of regulated computerized system validation required by government agencies like the FDA and the lifecycle of getting a product approved.
References:
- Glossary Of Computerized System And Software Development Terminology, 1996 (Note: This document is reference material for investigators and other FDA personnel. The document does not bind FDA, and does not confer any rights, privileges, benefits, or immunities for or on any person(s).)
- 21 CFR Part 11, electronic records; electronic signature rule, 20 March 1997, effective 20 August 1997.
- Society of Quality Assurance Virtual Training: Basic Computer System Validation training in 2023, Link.
- Regina Bailey, Steps of the Scientific Method. Updated August 16, 2024.
- FDA Part 11 Guidance Document on Scope and Application, 2003.
- Richard M. Siconolfi, MS, and Suzanne Bishop, MA, RAMP (Risk Assessment and Management Process): An Approach to Risk-Based Computer System Validation and Part 11 Compliance. Drug Information Journal, Vol. 41, pp. 69–79, 2007 • 0092-8615/2007.
- Computer Software Assurance for Production and Quality System Software, Draft Guidance for Industry and Food and Drug Administration, September 2022.
About The Author:
 Richie Siconolfi earned a BS in biology (Bethany College, Bethany, WV) and MS degree in toxicology (University of Cincinnati College of Medicine, Cincinnati). He has worked for The Standard Oil Co., Gulf Oil Co., Sherex Chemical Co., and the Procter & Gamble Co. Currently, Richie is a consultant in computer system validation, Part 11 compliance, data integrity, and software vendor audits (“The Validation Specialist”, Richard M Siconolfi, LLC). Richie is a co-founder of the Society of Quality Assurance and was elected president in 1990. He is a member of the Beyond Compliance Specialty Section, Computer Validation IT Compliance Specialty Section, and Program Committee. Richie also is a member of Research Quality Assurance’s IT Committee and Drug Information Association’s GCP/QA community. The Research Quality Assurance professional society appointed Richie to fellow in 2014.
Richie Siconolfi earned a BS in biology (Bethany College, Bethany, WV) and MS degree in toxicology (University of Cincinnati College of Medicine, Cincinnati). He has worked for The Standard Oil Co., Gulf Oil Co., Sherex Chemical Co., and the Procter & Gamble Co. Currently, Richie is a consultant in computer system validation, Part 11 compliance, data integrity, and software vendor audits (“The Validation Specialist”, Richard M Siconolfi, LLC). Richie is a co-founder of the Society of Quality Assurance and was elected president in 1990. He is a member of the Beyond Compliance Specialty Section, Computer Validation IT Compliance Specialty Section, and Program Committee. Richie also is a member of Research Quality Assurance’s IT Committee and Drug Information Association’s GCP/QA community. The Research Quality Assurance professional society appointed Richie to fellow in 2014.